Molecular Epidemiology of Infectious Diseases - computer practical: Pangenome analysis
Pangenome analysis
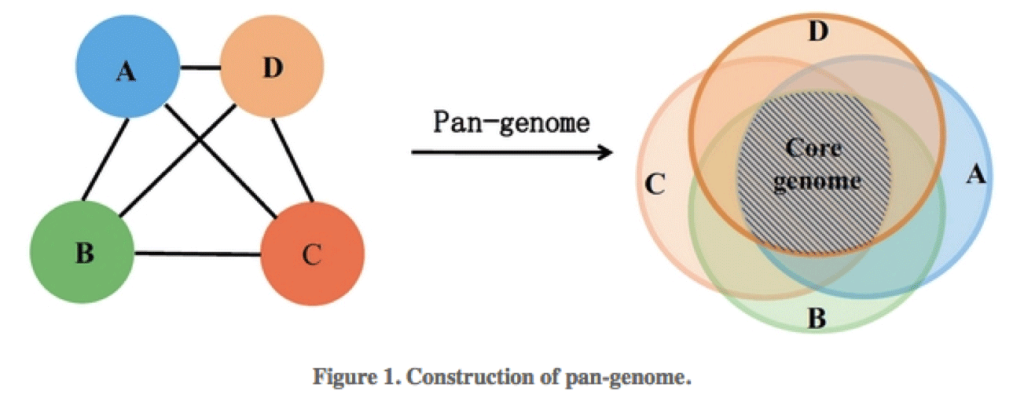
The microbial pangenome is the union of genes shared by genomes of interest. The core genome is the intersection of genes of these genomes, thus core genes are genes present in all strains. The accessory genome (also: variable, flexible, dispensable genome) refers to genes not present in all strains of a species. These include genes present in two or more strains or even genes unique to a single strain. Acquired antibiotic resistance genes are typically genes of the accessory genome.
Find more information of the pangenome here
Extract proteins RecB, RubA and DnaA from two genomes.
For this, we need to find if the genes are present in the annotated files.
Challenge: How can I extract genes from the file with the genes?
Hints:
grep -A 10 [gene] *faa gives you the first ten line after the gene name was foundSolution
$ cd annnotation $ grep -A 10 'recB' */*faa
Note for each genome if the gene has been found or not. Mark them as present (1) in this file, else mark the gene that differs or is absent as 0.
Pangenome analysis
Comparing every gene of every genome to every other gene in the dataset is an enourmous task, and takes a long time even if automated. Roary is a pipeline to determine genes of the core and pangenome. It takes a few short-cuts such as clustering instead of pair-wise alignment and can perform this task in a relatively short time frame. An excellent step-by-step tutorial can be found here
First we need to put the gff files generated by prokka into a separate folder
$ cd
$ mkdir ~/dc_workshop/results/annotation/gff_files
$ mv ~/dc_workshop/results/annotation/*/*gff ~/dc_workshop/results/annotation/gff_files
You can start a session and give it a descriptive name:
$ screen -S pangenome
This creates a session with the name ‘pangenome’
As you work, this session will stay active until you close it. Even if you log out or work on something else, the jobs you start in this session will run until completion.
then we can go to the gff directory and start roary
$ cd ~/dc_workshop/results/annotation/gff_files
$ roary *.gff -r -f ~/dc_workshop/results/pangenome/
You can detach from a session by pressing control + a followed by d (for detach) on your keyboard.
You can now safely work on something else. The process will keep on running. If you want to check on it, you can restore the session with
screen -r pangenome
Discussion: Open or closed pangenome?
After roary finished, have a look at the summary file. How many core and pangenome genes are there? Visit the definition of an open and closed pangenome here and download and view the file “Rplots.pdf”. Has M. tuberculosis a closed or an open pangenome?
Visualization
Some genes are present in all genomes, some are present in some and absent in others. Data on presence and absence of genes was collected in a matrix called gene_presence_absence.csv. Clustering of this information was used to build a tree (available as accessory_binary_genes.fa.newick). As a next step we are going to visualize this clustering.
Challenge: Which isolates are related based on gene presence/absence??
Copy accessory_binary_genes.fa.newick and gene_presence_absence.csv to your own computer. Open phandango in Chrome, drop file accessory_binary_genes.fa.newick and then file gene_absence_presence.csv. Make three groups out of the 7 isolates.
filezilla or scpcan be used to copy the files from the machine to your own computer
Solution
3 groups: .. .. ..